Maurilio Sampaolesi, Stefan Janssens & Robin Duelen
Publication: Expert Opinion on Therapeutic Targets vol. 27, 2023, issue 2
Abstract
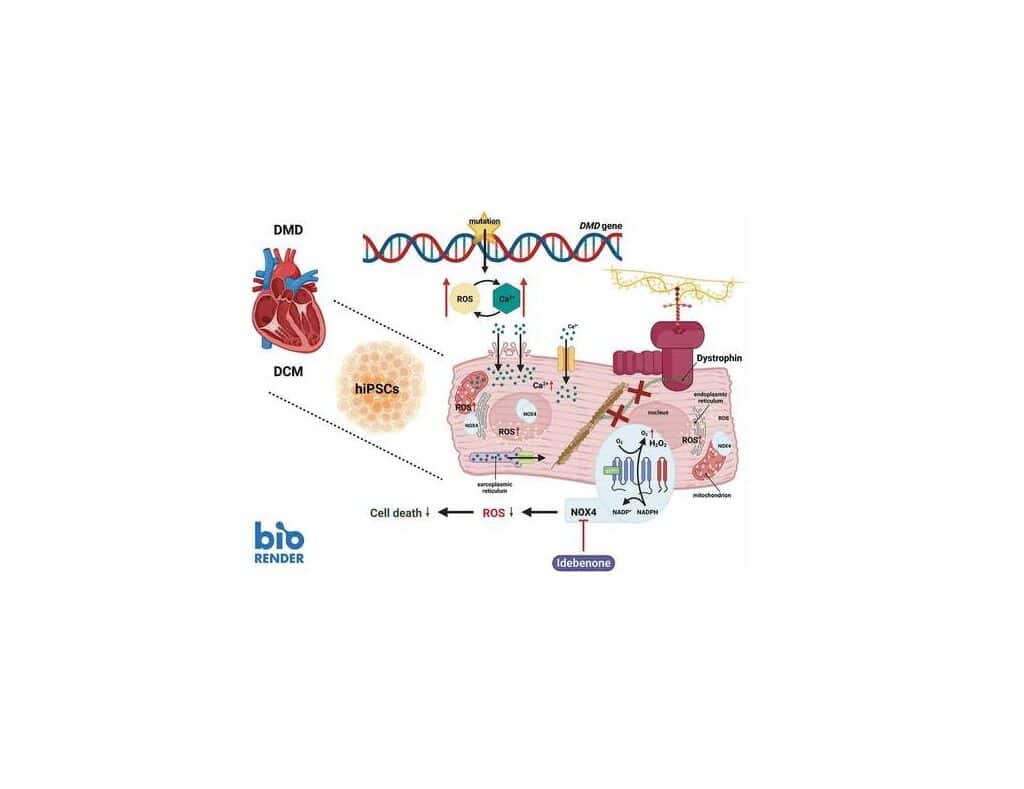
Duchenne muscular dystrophy is a pediatric muscle disorder, affecting approximately 1 in every 5000 male births worldwide. DMD is caused by mutations in the X-linked DMD gene, resulting in the absence of the functional form of its major protein product Dystrophin. The absence of Dystrophin results in several detrimental downstream effects, such as disruption of calcium (Ca2+) homeostasis, oxidative stress, inflammation, and mitochondrial dysfunction. Ultimately, these impairments make cardiac myocytes extremely vulnerable for degeneration, necrosis and thereby contributing to the progressive phase of cardiac fibrosis.
Adverse myocardial remodeling and chronic cardiomyopathy are major causes of morbidity and early mortality of most DMD patients. Initially, left ventricular (LV) dilation and hypertrophy develop to cope with the higher muscle wall stress, induced by pressure/volume overload. Subsequently, rhythm abnormalities (mainly supraventricular arrhythmias) and remodeling of the heart architecture occur that progress to dilated cardiomyopathy (DCM). In particular, repetitive mechanical stress causes apoptosis and myocardial fibrotic deposition. Eventually, heart failure and arrhythmias will develop as the disease progresses. Here, we focus on the increased oxidative stress as a major contributor to the development and progression of heart failure and more specifically in Duchenne cardiomyopathy.
Read the article.
